We are giving a glimpse at a subset of the powerful and expansive capabilities of the Isomorphic Labs Drug Design Engine (IsoDDE), a unified computational drug-design system, progressing beyond AlphaFold 3 (AF3) in its predictive accuracy and introducing new capabilities which bridge the gap between structure prediction and real-world drug discovery.
We demonstrate that our IsoDDE more than doubles the accuracy of AlphaFold 3 on a challenging protein-ligand structure prediction generalisation benchmark, predicts small molecule binding-affinities with accuracies that exceed gold-standard physics-based methods at a fraction of the time and cost, and is able to accurately identify novel binding pockets on target proteins using only the amino acid sequence as input.
IsoDDE offers a scalable foundation for AI drug design, providing the predictive fidelity required to navigate novel biological systems with unprecedented accuracy.
Since our release of AlphaFold 3 in 2024 together with Google DeepMind, the field of AI drug discovery has moved at an extraordinary pace. Whilst AlphaFold 3 delivered a dramatic leap in performance from previous generations of structure prediction models, a key challenge remained: understanding biomolecular structures alone was not sufficient for unlocking real-world drug discovery programs in silico (on a computer).
Progress in rational drug design - vital for solving human disease - requires highly accurate predictive models, across an expansive range of biochemical properties and interactions, that are able to work in concert with one another. Crucially, with so much of biological and chemical space still unexplored, these models need the ability to generalise their predictive power beyond their training sets to novel, unseen systems.
As we continue to address these challenges, we are excited to introduce the Isomorphic Labs Drug Design Engine (IsoDDE), and to preview a subset of IsoDDE's capabilities below and in our technical report.

Accurately predicting the structure of biomolecules and how they interact remains a crucial capability for rational drug design. Many critical downstream tasks are unlocked by being able to accurately model the small nuances in a protein’s geometry - whether understanding the impact of disease-causing mutations, or predicting which molecules will bind to a target protein.
AlphaFold 3 transformed protein-ligand structure prediction at the time of its release and the freely available AlphaFold Protein Database accelerated science on a scale that was previously unimaginable. To date, it has been used by over 3 million researchers in more than 190 countries.
Benchmarks have subsequently revealed that there remained a gap in accuracy for structures that were dissimilar to the examples AlphaFold 3 had been trained on. In other words, that it can struggle to generalise to unexplored regions of biomolecular space where some of the biggest challenges and opportunities in drug discovery lie.
IsoDDE demonstrates a step change in the ability to generalise to protein-ligand structures that are highly dissimilar to those in its training set.
On the 'Runs N' Poses' benchmark (Škrinjar et al. 2025) - designed specifically to test generalisation to novel pockets and ligands - IsoDDE more than doubles the accuracy of AlphaFold 3 on the most difficult systems.

In the report, we demonstrate through several examples that we can successfully model complex, out-of-distribution events such as induced fits (where a protein adapts its shape to accommodate a bound ligand) and the opening of cryptic pockets (those hidden in the absence of a bound ligand) - critical biological mechanisms - even when these systems are distant from the training sets of such models.
But small molecules (like aspirin) are only one piece of the puzzle. As therapeutic modalities expand toward complex biologics (like insulin), the ability to accurately model antibody-antigen interfaces is paramount.
IsoDDE provides a step change in accuracy for this domain. It outperforms AlphaFold 3 by 2.3x and Boltz-2 by 19.8x in the high-fidelity regime (DockQ > 0.8) on a challenging, novel antibody-antigen test set.
Crucially, IsoDDE shows remarkable performance on the CDR-H3 loop - the most variable and difficult part of an antibody to predict - effectively unlocking new possibilities for de novo antibody design.

Knowing the 3D structure of a biochemical system is only the first step; effective drug optimisation requires knowing how strongly a molecule will bind to its target.
Traditional approaches are either limited to chemical space similar to the training data or by their high computational cost and difficulty of execution (e.g., physics-based approaches). Deep-learning based methods have more recently emerged that bring new speed to this task, but still lag behind physics-based approaches for accuracy.
IsoDDE surpasses all deep-learning methods by a considerable margin on three public benchmarks - FEP+ 4, OpenFE, and the recent CASP16 blind binding affinity prediction task.
In fact, remarkably, IsoDDE can surpass the performance of physics-based methods such as FEP, despite the fact that these require grounding in experimental crystal structures and IsoDDE does not.
By delivering highly accurate binding affinity predictions at speed, IsoDDE allows researchers to rapidly rank and optimise potential molecules across diverse chemical series during drug design programs.

The ability to identify all of the potential pockets on a protein, in the absence of a known ligand, unlocks a number of unique opportunities. Whether dealing with a first-in-class drug discovery target lacking structural annotation or pursuing a novel way to modulate a well-studied protein, a general pocket identification capability can be used to reveal the full set of possible mechanisms of action to pursue for molecular design.
IsoDDE exhibits the capability to identify novel, ligandable pockets even in the absence of a known ligand and far from the model’s training set. This capacity for ‘blind’ pocket identification demonstrates performance levels approaching experimental techniques like fragment-soaking which require large investments in time, significant cost and real-world experimental work. In comparison, IsoDDE runs on a computer in a matter of seconds.
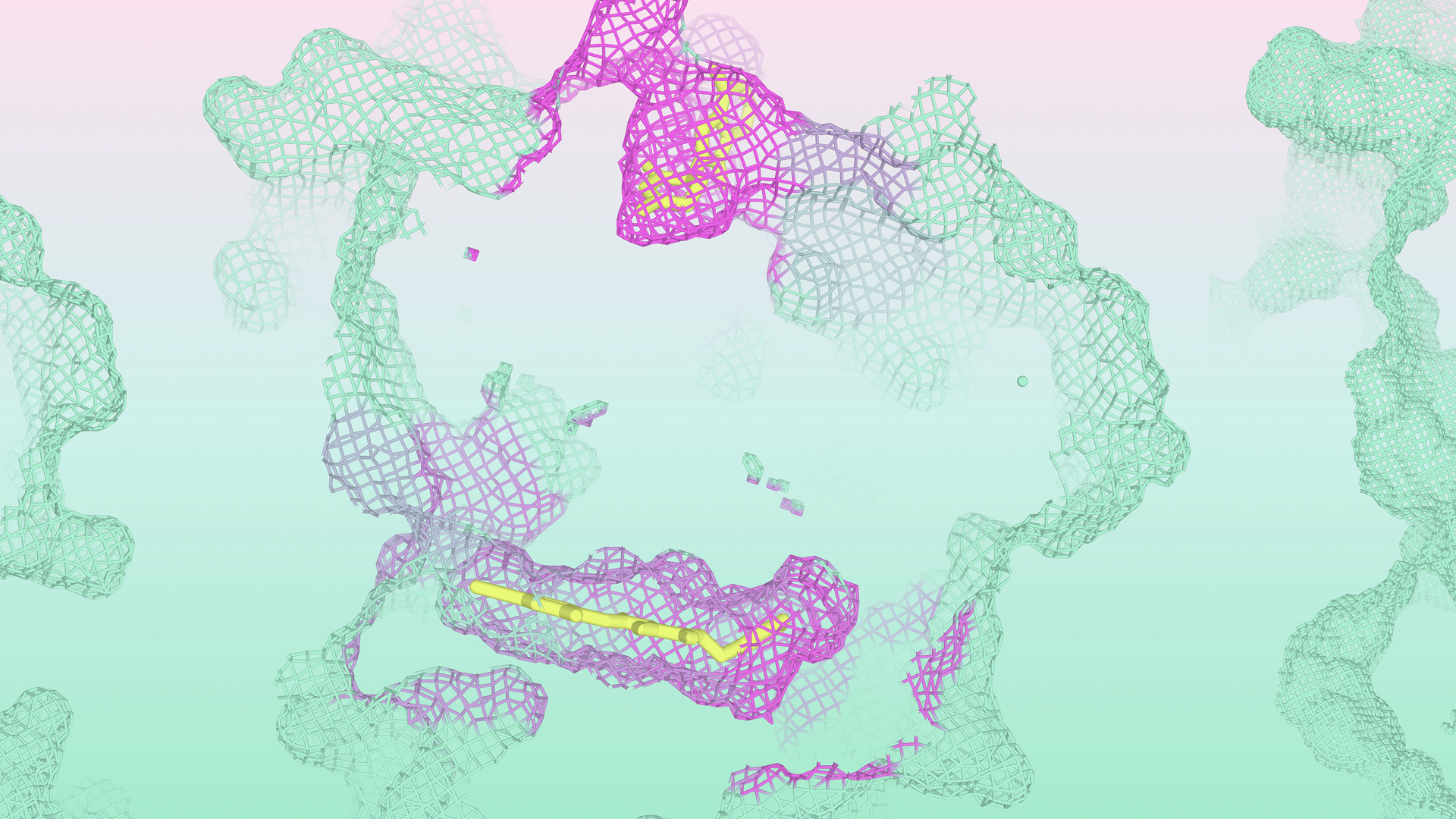
We can see the power of this capability in the example of cereblon - a substrate receptor for the CRL4 E3 ligase complex - which plays a key role in tagging damaged or misfolded proteins for proteasomal degradation. For the last 15 years, it was believed that there was one principal way to drug cereblon: through the classic thalidomide-binding pocket. However, a recent study (Dippon et al. 2026) experimentally discovered a novel binding pocket that was both allosteric (away from the traditional binding site) and cryptic (hidden in the absence of a binding ligand).
IsoDDE was able to recapitulate the discovery of this pocket, predicting the location of both the known and the novel cryptic sites using only the sequence of cereblon as input, without specifying the identity of the ligands. Further, once the ligands were specified, IsoDDE was able to correctly fold them into their respective pockets in the correct orientation.

IsoDDE represents a leap forward in accuracy and capability, bringing deeper understanding to the molecular machines that make up the human body, and advancing the process of designing drugs to modulate them.
Our dedicated drug design teams at Isomorphic Labs are using these capabilities every day across our programs – to understand unseen structures, identify uncharacterised pockets, and create novel chemical matter in the pursuit of new medicines for patients.
We look forward to continuing to push the frontiers of in silico drug design and bringing our new, more powerful capabilities to bear on historically challenging drug targets.
We thank our friends at Google DeepMind for productive discussions and collaboration.
Please use the following citation if you reference this work: